DataLabb: HTSC-struktur
Introduktion
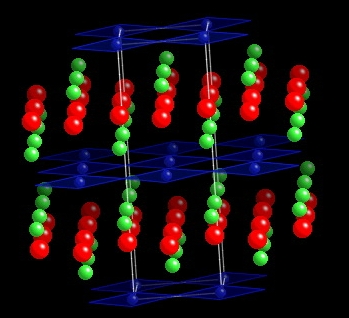
År 1986 upptäckte Bednorz och Müller supraledning vid rekordhöga temperaturer in en kopparoxid, La2CuO4, där en del av lantanet (3+) ersättes av strontium (2+) för att dopa CuO2-planen med hål.I La2CuO4 och i alla de hundratals högtemperaturledare som har uptäckts sedan dess finns det skickt av CuO2-plan, där varje syre-atom länkar två kopparatomer och där varje kopparatom har ett fyrkant av syre-atomer kring sig. Ofta finns det dessutom syre-atomer på något längre avstånd över och/eller under CuO2-planet, de så kallade apex-atomerna. CuO2-planen är ofta fyrkanta och kristallstrukturerna är ofta tetragonala. Ofta finns alla tre möjliga spegelplanen, så att de flesta tillhör rymdgruppen P4/mmm.
Uppgift
Handbook of Superconductivity (Academic Press 2000) av C.P.Poole innehåller en stor samling av kristallstrukurer av hög-temperatur supraledare. Jag ska dela ut olika supraledare till var och en av er, det vill säga en lista med gitterparametrer och en tabell med alla oberoende atompositioner i enhetscellen.- Starta CrystalMaker, och välj New Crystal under File menyn. Det första som ska matas in är rymdgruppen. Man kan göra det direkt, men det är bättre att välja Assist. De flesta av de här strukturerna tillhör det tetragonala kristallsystemet. Några är primitiva (P4), andra är centrerade (I4). (Förklara varför ytcentrerad är ekvivalent med kroppscentrerad för det tetragonala systemet.) Nu har du valt Bravaisgittret. Återstår valet av rymdgrupp med en lång pulldown menu. Ofta har de här strukturerna alla tre möjliga spegelplan, och då väljer man P4/mmm.
- Efter valet av rymdgrupp, matar man in data för de kristallparametrerna och för alla atompositioner som behövs. Varje grundämne har i programmet en default färg och jonstråle. I labeln kan man skriva vad som helst. Programmet genererar automatiskt de atompositioner som följer utav rymdgruppens symmetri.
- När alla atompostioner har matats in, kan man välja Plot. I ett fönster ser man en massa bollar som man kan vrida på med musrörelser. Under Model menyn kan man välja Space Filling eller Ball & Stick, men man ser inga sticks.
- Bindningar behövs för att se någon struktur i alla bollar. De måste först definieras under Edit-menyn i Bond Specs. Välje bindningar mellan koppar och syre på ett max-avstånd av 3 Ångström, och knäppa på Add.
- Nu visar Ball & Stick bindningar, men bara bland atompar i enhetscellen. Välja Set Range under Transformmenyn för att lägga till (delar av) celler i a eller b-riktningen.
- Välj nu Model Options under Model-menyn för att kunna visa polyedrar. Efter Cu står antagligen fortfarande ett kryss. Ändra den till en tilltalande polyeder. Syre-atomerna kan man ändra till bollar eventuellt med bindningar.
- Mät nu med avståndsverktyget de kortaste Cu-O avstånden.
- Välj lämplig Range och synvinkel för att skriva ut en tydlig bild av kristallstrukturen. (I Model Options kan man ändra den svarta bakgrunden.)
- Man kan leka vidare. Till exempel definiera ett hkl-plan, dra det genom strukturen, och ta bort atomer under eller över planet. Eller man kan titta på en kluster kring en viss tungmetallatom.
- Lägg in en del information (ämne, Tc, referens) från Poole i Notebookfönstret.
Diffraktion
Välj nu Diffraction Pattern under Transform-menyn. Det ska starta programmmet CrystalDiffract.- Vilken information ger positionen av den första peaken i diffraktionsmönstret?
- Varför är 010-intensiteten så mycket mindre än 020-intensiteten?
- 102 och 103 brukar ha stor intensitet is dessa strukturer. Välj den starkaste av dessa och rita Crystal Plane med dessa hkl-indices i CrystalMaker. Välj en orientation så att men ser planet från sidan, och flytta den i sidled med planförflyttningverktyget. Förklara varför dessa plan har så stark Braggreflektion. Bifog figurer.
- Jämför intensiteterna för röntgen och för neutrondiffraktion. Varför är de så olika?